RESEARCH APPROACH
Cu-directed hydroxylation of C-H bonds
In organic synthesis, an elegant way to overcome the selectivity issues associated with the formation of radicals is the use of directing groups (DG). In collaboration with Prof. Phil Baran, we studied the mechanism by which Cu promotes the intramolecular g-hydroxylation of sp3 C-H bonds, which allowed a redesign of the oxidation conditions to use cheaper reagents and milder conditions with improved overall yields (J. Org. Chem. 2017, 82, 7887). Our lab later discovered that other sp2 and sp3 C-H substrates could also be oxidized using imino-pyridine DGs, Cu and H2O2 (Inorg. Chem. 2019, 58, 7584). In both cases, we proposed the formation of a mononuclear CuIIOOH core prior to C-H hydroxylation, suggesting that mononuclear Cu/O2 species could be formed in Cu-dependent mononooxygenases. New research directions include the utilization of DGs with varying denticity and using this approach to develop synthetically useful transformations (e.g. enantioselective hydroxylations).

Structure, spectroscopy and reactivity of 3d metal complexes bearing bidentate redox-active ligands with tunable H-bonding donors
An alternative approach to avoid the formation of radical species in 3d metal-catalyzed organic synthesis is the use of redox-active ligands. In 2018, we reported that Cu complexes bearing bidentate redox-active ligands with tunable H-bonding groups catalyzed the aerobic oxidation of alcohols under mild conditions (J. Am. Chem. Soc., 2018, 140, 16625). We found that these unique ligands donated e− and stabilized Cu/O2 species via H-bonding, and that the mechanism of alcohol oxidation was unprecedented for galactose-oxidase model systems. New research directions include the systematic study of these H-bonding interactions, the development of catalysts for biologically relevant reactions and the characterization and reactivity of “high-valent” metal complexes.

Monooxygenase-like reactivity of 3d metal complexes bearing tridentate redox-active ligands with tunable H-bonding donors
Inspired by the active site of LPMO, we have recently reported the synthesis of a CuOH complex bearing an analogous tridentate ligand. This CuOH core was oxidized to three molecular oxidation states and that these “high-valent” species perform two consecutive H abstractions from phenolic substrates via H transfer (J. Am. Chem. Soc., 2020, 142, 12265). We will utilize these ligands to study intramolecular H-bonding interactions (between the anion and the H-bond donor), to develop metal complexes capable of functionalizing C-H bonds (hydroxylations, C-N bond formation, etc.), and to synthesize and characterize Cu-oxyl species (the “lost” oxidant).

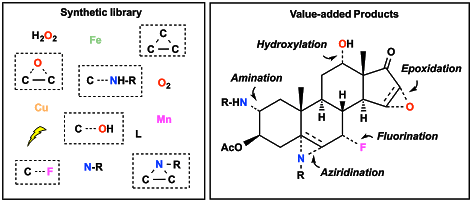
New Bio-Inspired Catalysts for Practical Organic Synthesis
Enzymatic catalytic transformations have inspired synthetic chemists to design simplified catalytic systems that could emulate and/or overcome these natural processes. Many researchers have expended efforts to explore new routes in the conversion of inert C-H bonds to value-added products. This part of the scientific project will focus on the development of catalysts based on the methodical design of first row metal complexes for the catalytic transformation of C-H and C=C bonds to selective C-O bond formation (hydroxylations, cis-dihydroxylatyion and epoxidation), C-N formation (aminations, aziridinations), C-C formation (cyclopropanation), and C-X halogenations (X = F, Cl, Br, I). Our efforts will concentrate on building a library of synthetic tools for cheap, environmentally sustainable, chemo- and stereoselective chemical transformations of complex molecules.